Amiloidose hereditária ATTR: uma doença familiar progressiva e com risco de morte1-3
O que é amiloidose hATTR?
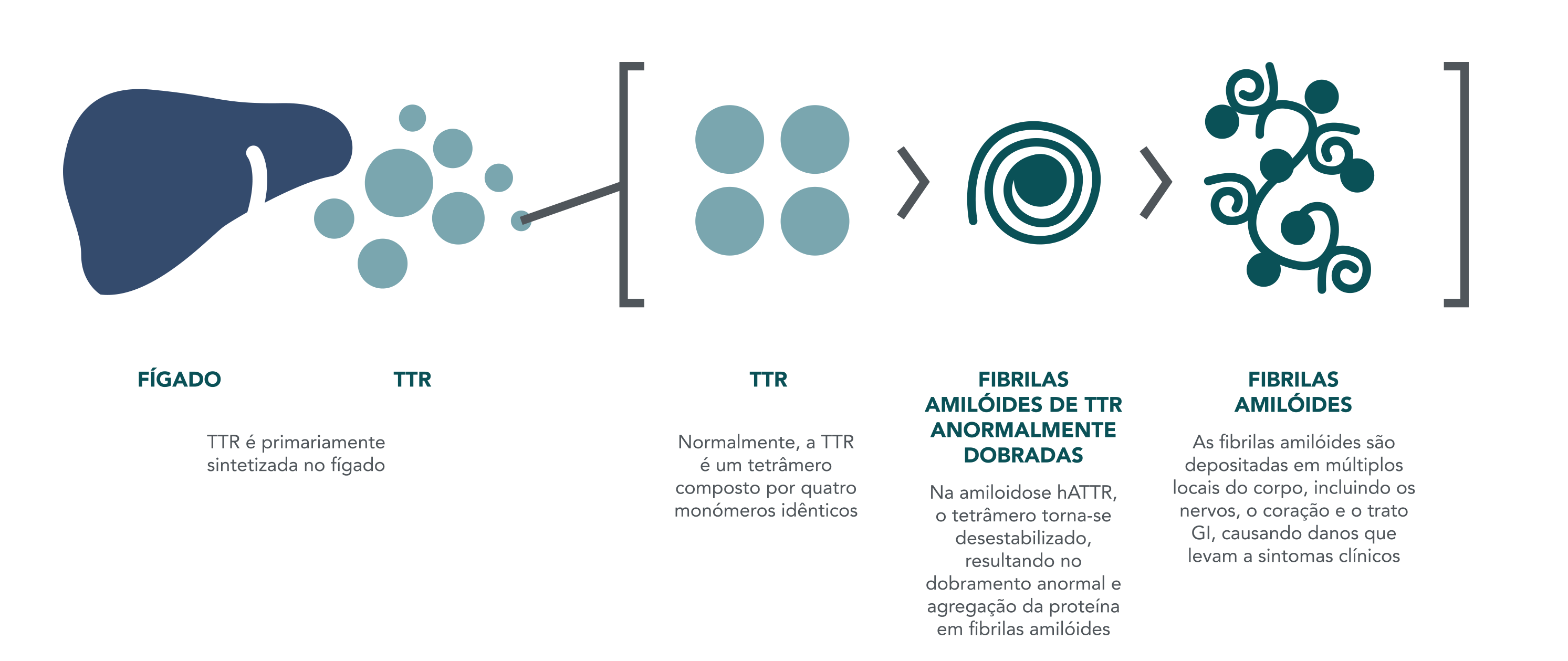
A amiloidose hATTR é uma doença autossômica dominante causada por uma mutação no gene transtirretina (TTR) que resulta no acúmulo de proteínas TTR com enovelamento anormal como fibrilas amiloides em diversos locais, incluindo nervos, coração e trato GI.2,4,5
Formação das fibrilas amiloides2,4-6
Referências:
- Adams D, Coelho T, Obici L, et al. Rapid progression of familial amyloidotic polyneuropathy: a multinational natural history study. Neurology. 2015;85(8):675-682.
- Hanna M. Novel drugs targeting transthyretin amyloidosis. Curr Heart Fail Rep. 2014;11(1):50-57.
- Mohty D, Damy T, Cosnay P, et al. Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis. 2013;106(10):528-540.
- Damy T, Judge DP, Kristen AV, et al. Cardiac findings and events observed in an open-label clinical trial of tafamidis in patients with non-Val30Met and non-Val122lle hereditary transthyretin amyloidosis. J Cardiovasc Transl Res. 2015;8(2):117-127.
- Hawkins PN, Ando Y, Dispenzeri A, et al. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625-638.
- Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease-modifying treatments. J Neurol Neurosurg Psychiatry. 2015;86(9):1036-1043.
- Ando Y, Coelho T, Berk JL, et al. Guidelines of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31.
- Rapezzi C, Quarta CC, Obici L, et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J. 2013;34(7):520-528.
- Adams D, Gonzalez-Duarte A, O’Riordan W, et al., an investigational RNAi therapeutic for the treatment of hereditary ATTR amyloidosis with polyneuropathy: baseline demographics from the phase 3 APOLLO study. In: The XVth International Symposium on Amyloidosis. Uppsala, Sweden: ISA International Society of Amyloidosis; July 3-7, 2016. PA 82.
- Coelho T, Maurer MS, Suhr OB. THAOS—The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29(1):63-76.
- Castaño A, Drachman BM, Judge D, et al. Natural history and therapy of TTR-cardiac amyloidosis: emerging disease-modifying therapies from organ transplantation to stabilizer and silencer drugs. Heart Fail Rev. 2015;20(2):163-178.